Council of Canadian Academies
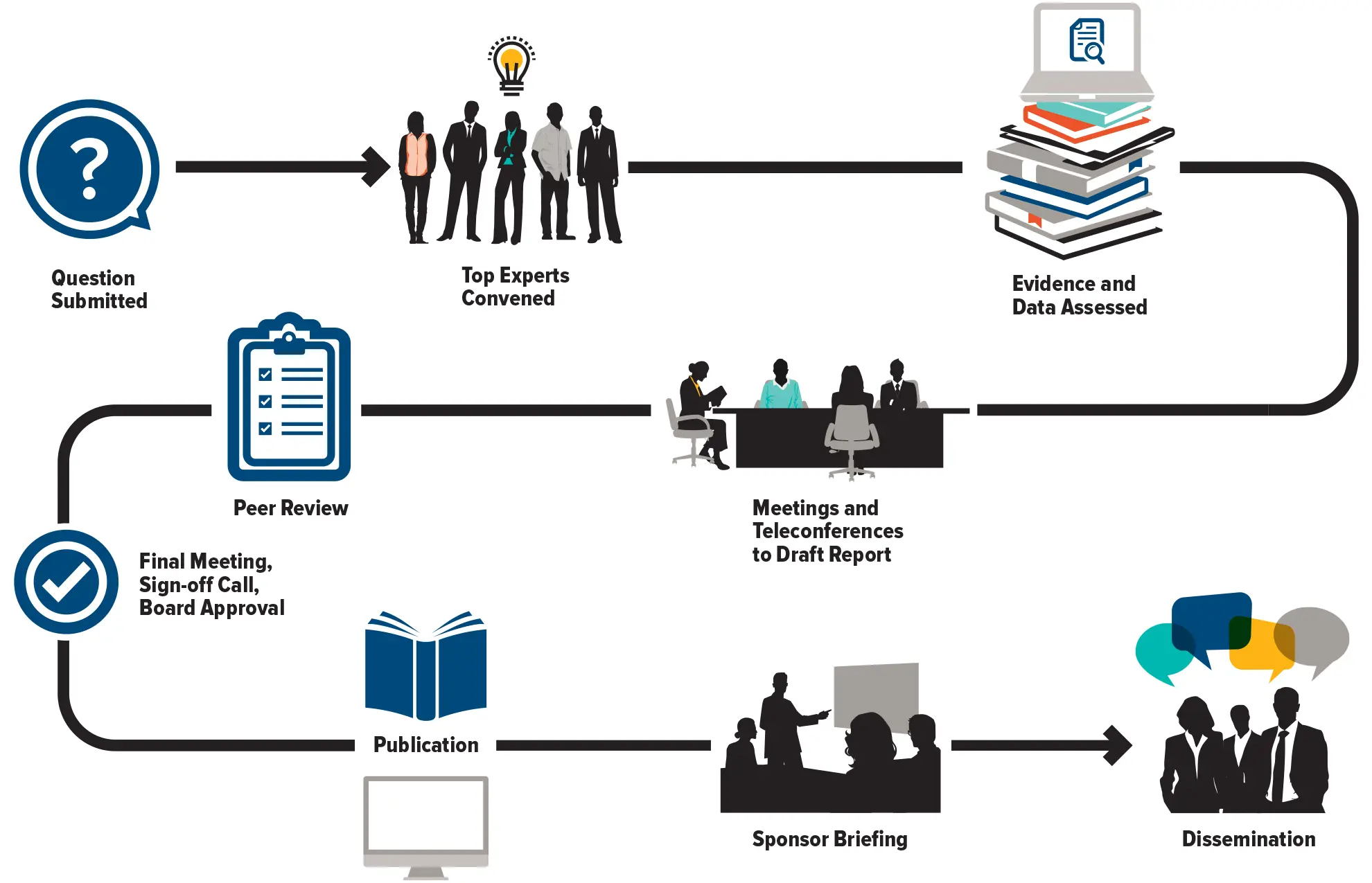
The Council of Canadian Academies (CCA) convenes independent panels of leading experts to analyze and interpret the best available knowledge on issues of importance to Canadians. Our reports cover pressing issues in a range of policy areas—including health and life sciences, technology and innovation, the environment, and public safety—and provide a trusted source of reliable information widely used by decision-makers across governments, industry, academia, and civil society.